Research
1. Physical/Chemical Properties and Interdomain Dynamics Governing the Biological Functions of PICK1 in Substance Use Disorders (SUD)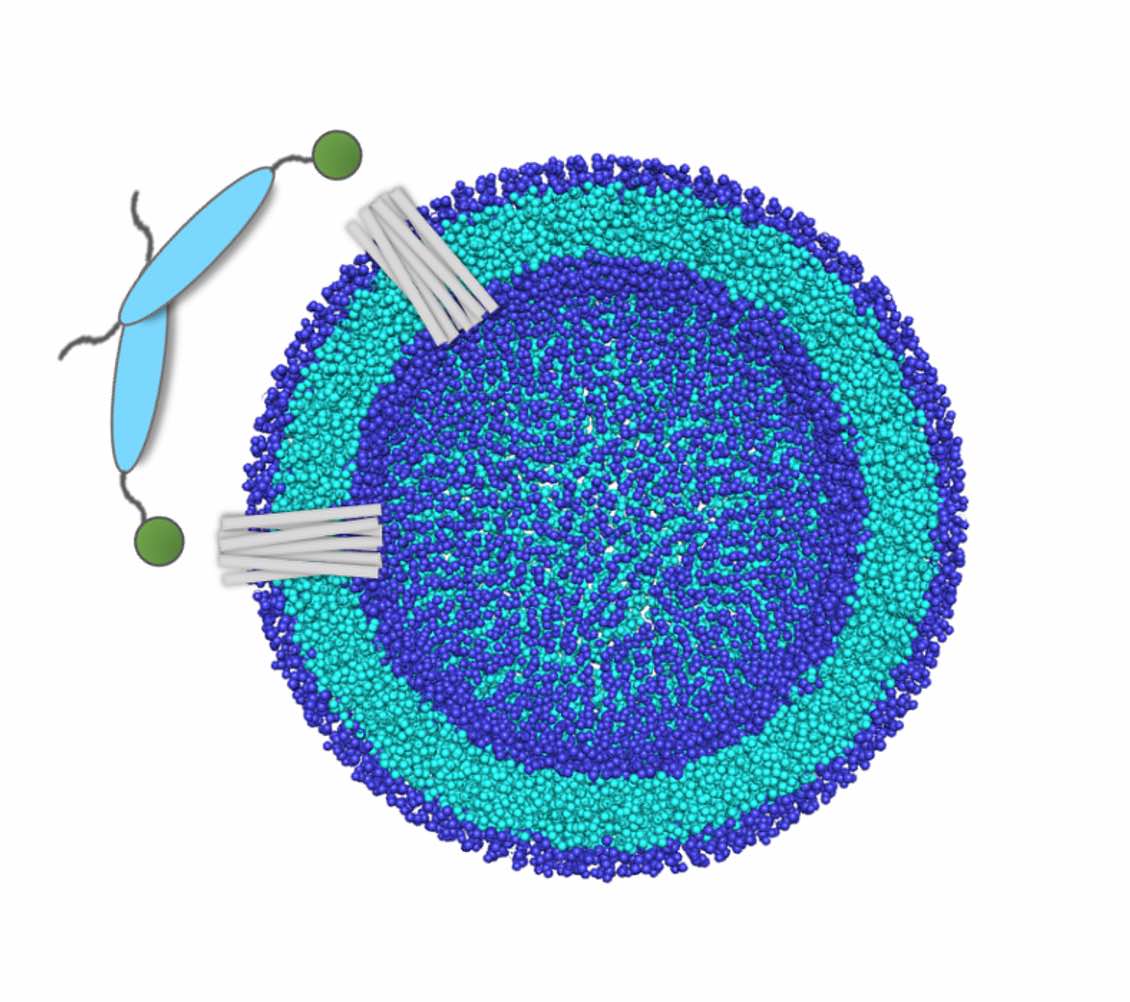
The PICK1 protein plays a critical role in regulating synaptic plasticity and neurotransmitter release and has been linked to several neurological and psychiatric disorders, such as SUD. Understanding the structural and functional mechanism of PICK1 is crucial to developing effective therapeutics for these conditions. However, the traditional experimental approaches for studying protein function are limited in their ability to provide a detailed understanding of protein interactions and mechanisms. Computational methods, such as AlphaFold, multiscale modeling, molecular dynamics simulations, and free energy calculations, have emerged as powerful tools to investigate protein-protein interactions and their functional mechanisms. Therefore, the use of computational methods to understand PICK1's functional mechanism has significant potential to uncover new insights into the protein's role in synaptic transmission and to aid in the development of novel therapeutics. We began by examining each component of the PICK1 protein. The PDZ domain belongs to a large protein family. We started by reviewing previous research on the PDZ domain [1] and then delved into the unique allosteric responses of the PICK1-PDZ domain to various ligands.[2,3] For BAR domains, we identified the key residues and the role of the kinks in determining their mechanical properties.[4] Concurrently, we studied how trimming specific regions of PICK1 impacts its biological functions.[5] Additionally, using SAXS data as a guide, we explored the interdomain dynamics of PICK1. This helped us understand the relationship between the PDZ and BAR domains, as well as the role of the flexible linker in these processes.[6,7]
[1]. Amy O. Stevens, Yi He* - Allosterism in the PDZ family, Int. J. Mol. Sci., 23(3):1454, 2022
[2]. Amy O. Stevens, Samuel Luo, Yi He* -Three binding conformations of BIO124 in the pocket of the PICK1 PDZ domain, Cells, 11(15), 2451, 2022
[3]. Amy O. Stevens, John Kazan, Banu Ozkan*, Yi He* - Investigating the allosteric response of the PICK1 PDZ domain to different ligands with all-atom simulations, Protein Sci., e4474, 2022
[4]. Shenghan Song, Tongtong Li, Taha Raad##, Amy O. Stevens, Yi He* - How key residues determine the mechanical responses of BAR dimer to external force, Curr. Protein Pept. Sci., 2023
[5]. Amy O. Stevens, Yi He* - Residue-level contact reveals modular domain interactions of PICK1 are driven by both electrostatic and hydrophobic forces, Front. Mol. Biosci., 7:6161352020, 2021
[6]. Tongtong Li, Laura I. Gil Pineda, Amy O. Stevens, Yi He* - Key factors regulating the inter-domain dynamics may contribute to the assembly of ASC, Biology, 12(6), 796, 2023
[7]. Amy O. Stevens, Yi He* - Structure ensemble reconstructions of the flexible PICK1 dimer using the SAXS data, Nucleic Acid Research, 2023 (In preparation)
2. CompuSulfono - AlphaFold guided drug/scaffold design using sulfono-γ-AApeptides
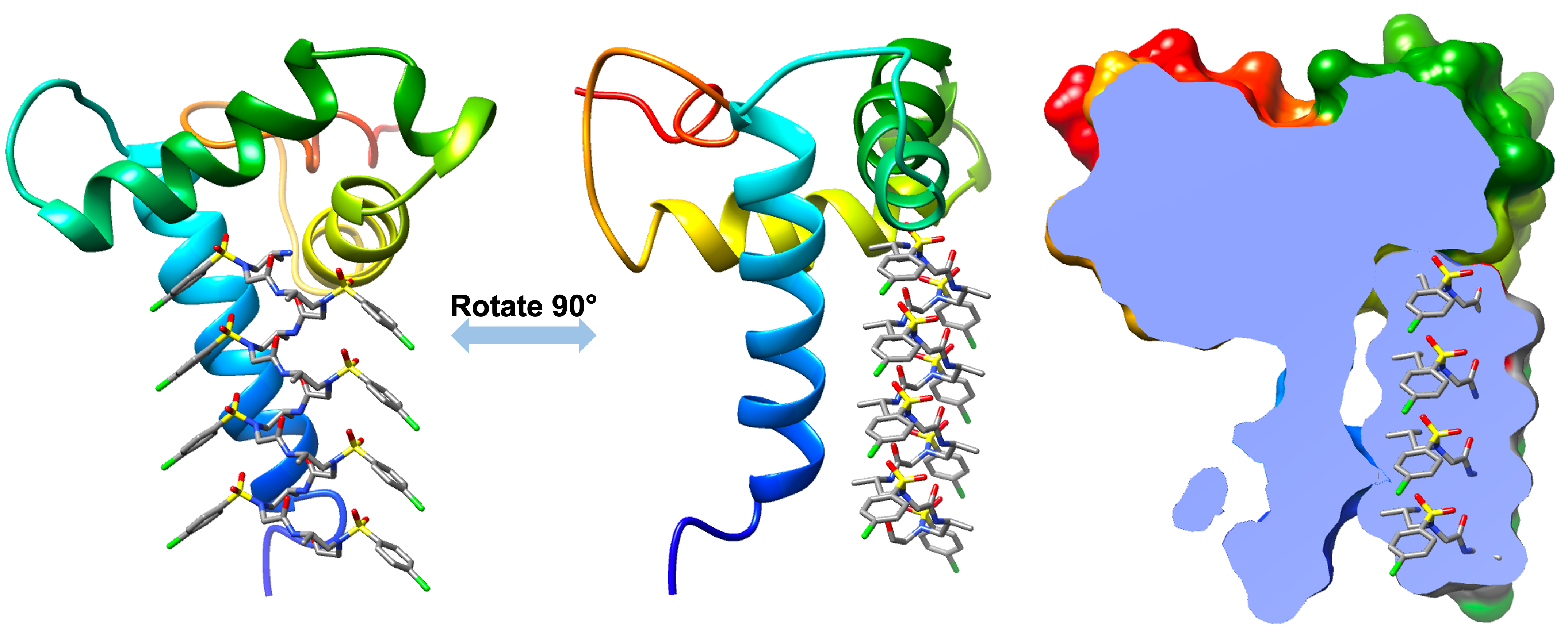
[1]. Jessica L. Binder, Joel Berendzen, Amy O. Stevens, Jian Wang, Yi He, Nikolay V. Dokholyan, & Tudor I. Oprea* - AlphaFold Models Illuminate Half of Dark Human Proteins, 74:102372, Curr. Opin. Struct. Biol., 2022
[2]. Amy O. Stevens, Yi He* - Evaluation of loop structures in AlphaFold 2 predictions, Biomolecules, 12(7), 985, 2022
[3. ]Catriona Gordon#, Emily Hendrix#, Yi He, Mark Walker* - A ColabFold-based computational pipeline to accurately predict RiPP complex structures, Biomolecules, 13(8), 1243, 2023
[4]. Emily Hendrix, Yi He* - Optimizing Ligand Design for PICK1 PDZ Inhibition: A Benchmark Analysis of AfDesign's Capabilities and Constraints, RSC Chemical Biology, 2023
[5]. Tongtong Li, Shenghan Song, Yi He* - Temporary Solution for Investigating γ-AApeptide Packing via Molecular Dynamics, Supramolecular Materials, 2023
4. Experimental data guided computational approaches to explore intrinsically disordered Proteins.
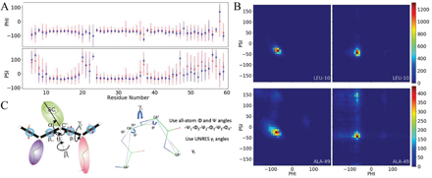 Intrinsically Disordered Proteins (IDPs) are a class of proteins that lack a well-defined three-dimensional structure under physiological conditions. These proteins play essential roles in a wide range of biological processes, including cell signaling and regulation, transcription, and translation. NMR spectroscopy is a powerful technique for studying IDPs, as it provides detailed information on the conformational ensemble of the protein. Multiscale computational approaches can integrate NMR data with molecular simulations to explore the conformational landscape of IDPs. These approaches use information from multiple scales, such as atomistic and coarse-grained simulations, to accurately model the dynamic behavior of IDPs. By combining experimental data with computational simulations, researchers can gain a more comprehensive understanding of the structure and function of IDPs and their interactions with other proteins and ligands. This can have important implications for drug discovery and the development of new therapies for diseases associated with IDPs, such as neurodegenerative diseases and cancer. We began by benchmarking various all-atom and coarse-grained force fields to gauge their performance.[1,2] We examined the binding and unbinding pathways of the protein complex system, p53 TAD2 - p300 Taz2, to pinpoint the crucial factors that determine the final binding structure of p53 TAD2, which is an Intrinsically Disordered Protein (IDP).[3-5] To understand the limitations of AlphaFold in predicting IDP ensembles, we directly compared the structural ensemble predicted by AlphaFold with computer-simulated ensembles and experimental data.[6] Surprisingly, in many instances, AlphaFold was able to generate probabilities for secondary structures.
Intrinsically Disordered Proteins (IDPs) are a class of proteins that lack a well-defined three-dimensional structure under physiological conditions. These proteins play essential roles in a wide range of biological processes, including cell signaling and regulation, transcription, and translation. NMR spectroscopy is a powerful technique for studying IDPs, as it provides detailed information on the conformational ensemble of the protein. Multiscale computational approaches can integrate NMR data with molecular simulations to explore the conformational landscape of IDPs. These approaches use information from multiple scales, such as atomistic and coarse-grained simulations, to accurately model the dynamic behavior of IDPs. By combining experimental data with computational simulations, researchers can gain a more comprehensive understanding of the structure and function of IDPs and their interactions with other proteins and ligands. This can have important implications for drug discovery and the development of new therapies for diseases associated with IDPs, such as neurodegenerative diseases and cancer. We began by benchmarking various all-atom and coarse-grained force fields to gauge their performance.[1,2] We examined the binding and unbinding pathways of the protein complex system, p53 TAD2 - p300 Taz2, to pinpoint the crucial factors that determine the final binding structure of p53 TAD2, which is an Intrinsically Disordered Protein (IDP).[3-5] To understand the limitations of AlphaFold in predicting IDP ensembles, we directly compared the structural ensemble predicted by AlphaFold with computer-simulated ensembles and experimental data.[6] Surprisingly, in many instances, AlphaFold was able to generate probabilities for secondary structures.[1]. Laura I. Gil##, Laurie N. Milko##, Yi He* - Performance of CHARMM36m with modified water model in simulating intrinsically disordered proteins: A case study, Biophysics Reports, 1, 2020, DOI:10.1007/s41048-020-00107-w
[2]. Tongtong Li, Emily Hendrix, Yi He* - Simple and effective conformational sampling strategy for intrinsically disordered proteins using UNRES web server, J. Chem. Phys. B, 2023
[3]. Tongtong Li, Amy O. Stevens, Laura I. Gil Pineda, Shenghan Song, Christabel A. Ameyaw Baah, Yi He* - Changes in structure and flexibility of p53 TAD2 upon binding to p300 Taz2, J. Theor. Comput. Chem., 2040007, 2020 DOI: 10.1142/S0219633620400076
[4]. Tongtong Li, Stefano Motta*, Amy O. Stevens, Shenghan Song, Emily Hendrix, Alessandro Pandini, Yi He* - Recognizing the binding pattern and dissociation pathways of p300 Taz2 - p53 TAD2 complex, J. Am. Chem. Soc. Au, 2(8) 1935–1945, 2022
[5]. Tongtong Li, Yi He* - Reveal the role of key residues guiding the binding process of p300 Taz2 and p53 TAD2, J. Chem. Theory Comput., 2023 (In preparation)
[6]. Tongtong Li, Yi He* - AlphaFold 2: Potential and Limitations in Probing Structural Ensembles of Intrinsically Disordered Proteins Compared to All-atom Simulations, Nat. Commun., 2023 (In preparation)
